Je rok 1982. Stanley Prusiner publikuje v časopise Science hypotézu, ktorá je väčšinou kolegov okamžite odmietnutá ako biologicky absurdná. Jeho tvrdenie: existuje infekčný patogén bez DNA, bez RNA, bez akejkoľvek genetickej informácie. Len proteín so zlým tvarom.
Väčšina vedcov bola presvedčená, že to je nemožné. Prusiner strávil ďalších pätnásť rokov dokazovaním, že majú pravdu len čiastočne — a v roku 1997 dostal Nobelovu cenu.
Prióny zmenili základný predpoklad biológie: že biologická informácia musí byť zakódovaná v génoch. Ukázalo sa, že niekedy stačí tvar.
Keď sa proteín stane infekčným
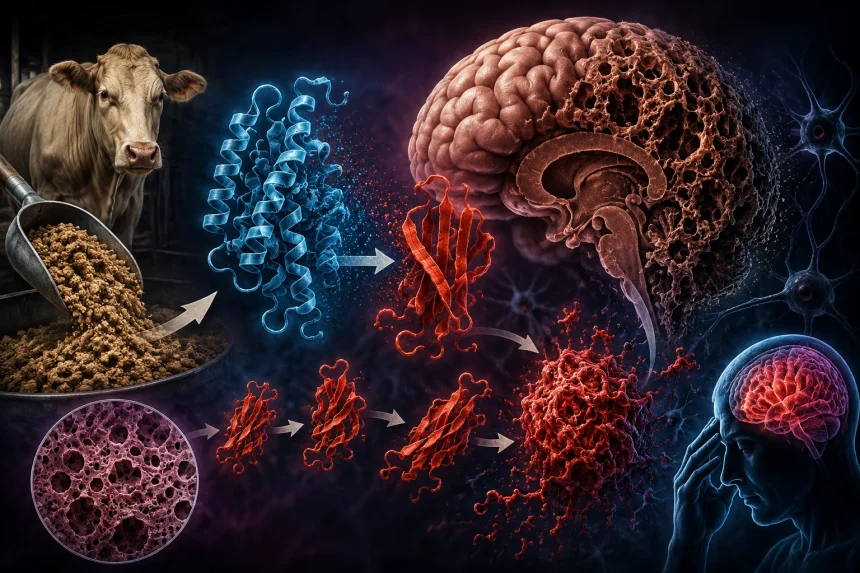
Slovo prión pochádza z anglického výrazu proteinaceous infectious particle – bielkovinová infekčná častica. Na rozdiel od vírusov či baktérií nejde o živý organizmus, ale o proteín s nesprávne poskladanou priestorovou štruktúrou.
V nervovom systéme cicavcov sa prirodzene nachádza proteín označovaný ako PrPC. Hoci jeho presná fyziologická úloha nie je úplne objasnená, uvažuje sa o úlohe v signalizácii a homeostáze neurónov. Za určitých okolností sa však môže zmeniť na patologickú formu označovanú ako PrPSc. K tejto premene môže dôjsť spontánne, v dôsledku genetickej mutácie alebo po kontakte s už existujúcim patologickým priónom.
Práve v tom spočíva jedinečnosť priónov: nenesú genetickú informáciu, ale informáciu o svojom tvare.
Molekulárne origami
Predstavte si list papiera. Chemické zloženie zostáva rovnaké, no podľa spôsobu skladania môže vzniknúť lietadielko, loďka alebo žeriav. Pri proteínoch je situácia podobná. Normálny a patologický prión pozostávajú z rovnakých aminokyselín. Rozdiel je iba v ich trojrozmernom usporiadaní. Patologická forma je mimoriadne stabilná a zároveň dokáže prinútiť zdravé molekuly, aby prijali rovnaký chybný tvar.
Výsledkom je reťazová reakcia. Jeden nesprávne poskladaný proteín postupne premieňa ďalšie a ďalšie molekuly. V mozgu sa následne hromadia agregáty patologických proteínov a dochádza k postupnému poškodeniu neurónov. Mikroskopický obraz postihnutého mozgu pripomína špongiu plnú drobných otvorov. Odtiaľ pochádza názov skupiny ochorení spôsobených priónmi – transmisívne spongiformné encefalopatie.
Choroby, ktoré zmenili medicínu
Najznámejším priónovým ochorením človeka je Creutzfeldt-Jakobova choroba. Ide o zriedkavé, ale vždy smrteľné neurodegeneratívne ochorenie, ktoré sa prejavuje rýchlo postupujúcou stratou pamäti, poruchami koordinácie pohybu, zmenami správania a demenciou.
V 90. rokoch 20. storočia sa prióny dostali do centra pozornosti verejnosti v súvislosti s epidémiou bovinnej spongiformnej encefalopatie (BSE), známej ako „choroba šialených kráv“. Epidémia vznikla najmä v dôsledku kanibalizmu v rámci druhu – skrmovania mäsokostnej múčky vyrobenej z nervového tkaniva infikovaných zvierat. Recyklácia vlastného druhu v krmivovom reťazci umožnila patologickým priónom prenášať sa medzi jedincami, kde následne fungovali ako templát – vzorec, podľa ktorého sa normálne proteíny hostiteľa postupne preklápali do patologickej formy. Tento reťazový proces viedol po dlhej inkubačnej dobe (niekoľko rokov) k masívnej akumulácii poškodených proteínov v mozgu a následnej ťažkej neurodegenerácii.
Táto udalosť významne prispela k sprísneniu potravinovej bezpečnosti a veterinárnych kontrol na celom svete a stala sa varovným príbehom o rizikách recyklácie živočíšnych proteínov.
Prečo sú prióny také odolné?
Jednou z najpozoruhodnejších vlastností priónov je ich mimoriadna odolnosť. Keďže neobsahujú DNA ani RNA, mnohé postupy určené na ničenie mikroorganizmov na ne nefungujú. Prióny vykazujú mimoriadnu odolnosť voči teplu a mnohým štandardným sterilizačným postupom. Ich úplná inaktivácia si vyžaduje podstatne prísnejšie podmienky než pri väčšine baktérií a vírusov. Táto vlastnosť si vyžiadala špeciálne sterilizačné protokoly pre nástroje, ktoré prichádzajú do kontaktu s nervovým tkanivom. Imunitný systém proti nim takmer nebojuje– pretože ide o modifikovanú verziu vlastného proteínu, organizmus ho často nerozpozná ako cudzieho, neidentifikuje ho ako cudzí patogén.
Vzhľadom na extrémnu odolnosť priónov pri bežnej sterilizácii a spracovaní surovín sa recyklácia biologických tkanív, najmä tých nervových, stala kritickým bodom, ktorý umožnil nekontrolované šírenie týchto patogénov v chovoch hospodárskych zvierat.
Infobox: Prióny a prion-like mechanizmy v bežných ochoreniach
Prióny nie sú jediným prípadom, keď nesprávne poskladané proteíny spôsobujú problémy. Podobný mechanizmus (tzv. prion-like) sa dnes spája aj s bežnými neurodegeneratívnymi ochoreniami:
Alzheimerova choroba — beta-amyloid a tau proteín sa môžu šíriť mozgom ako „semienka“. Výsledkom je postupná strata pamäti, orientácie, schopnosti plánovať a samostatne sa rozhodovať. Choroba ničí najmä kortex a hipokampus, čo vedie k úplnej závislosti pacienta od okolia v neskorších štádiách.
Parkinsonova choroba — alfa-synukleín vytvára toxické agregáty (Lewyho telieska) v dopaminergných neurónoch. Prejavuje sa trasením, stuhnutosťou, spomalením pohybu a neskôr aj poruchami kognície a depresiou. Postihnutí postupne strácajú schopnosť samostatného pohybu a kontroly nad telom.
ALS (amyotrofická laterálna skleróza) — proteín TDP-43 sa nesprávne skladá, agreguje sa a existujú dôkazy, že patologický TDP-43 môže vykazovať prion-like vlastnosti a šíriť sa medzi bunkami, čo vedie k postupnému odumieraniu motorických neurónov. Toto ochorenie postihuje najmä mladších dospelých a má veľmi rýchly priebeh.
Pochopenie týchto mechanizmov patrí medzi najperspektívnejšie smery súčasnej neurovedy. Niekoľko experimentálnych liekov cielených na zastavenie tohto „prion-like“ šírenia je už v klinických testoch.
Kľúč k pochopeniu neurodegeneratívnych ochorení?
Význam priónového výskumu, za ktorý získal Stanley B. Prusiner v roku 1997 Nobelovu cenu, dnes ďaleko presahuje samotné priónové ochorenia. Mnohé neurodegeneratívne choroby totiž vykazujú podobný mechanizmus – nesprávne poskladané proteíny vytvárajú toxické agregáty, ktoré sa šíria mozgom a poškodzujú neuróny.
Pri Alzheimerovej chorobe ide najmä o beta-amyloid a tau proteín, pri Parkinsonovej chorobe o alfa-synukleín. Niektorí vedci preto hovoria o tzv. prion-like mechanizmoch. Pochopenie týchto procesov patrí medzi najperspektívnejšie smery súčasnej neurovedy.
PrPSc agregáty môžu v budúcich konceptoch slúžiť ako ciele pre nanočastice navrhnuté na selektívne viazanie patologických konformácií proteínov. Experimentálne prístupy s magnetickými nanočasticami skúmajú možnosť lokálneho generovania tepla v ich okolí (hypertermia), ktoré by mohlo prispieť k destabilizácii týchto agregátov a ich následnej degradácii.
Budúcnosť terapie: Nanotechnológie a fyzikálna medicína
Hoci sú priónové ochorenia v súčasnosti nevyliečiteľné, moderná veda sa vydala cestou fyzikálnej dezintegrácie. Perspektívny prístup využíva cielenú magnetickú hypertermiu, ktorá stojí na troch pilieroch:
- Molekulárne navádzanie: Vývoj špecifických nanočastíc ozbrojených ligandami (napr. aptamérmi). Tie dokážu selektívne rozpoznávať a viazať sa výhradne na patologicky poskladané prióny (PrPSc), ktoré sa postupne zhlukujú do toxických agregátov. Práve tieto zhluky sú zodpovedné za postupné poškodzovanie neurónov, čo v konečnom dôsledku vedie k vzniku charakteristického „špongiovitého“ vzhľadu mozgového tkaniva. Zdravé proteíny (PrPC) tieto nanočastice ignorujú.
- Fyzikálne zosilnenie a „tepelný dezintegrátor“: Po aplikácii externého elektromagnetického poľa začnú tieto nanočastice pôsobiť ako lokálne antény. Premieňajú energiu poľa na teplo v bezprostrednom okolí priónového zhluku. Fyzikálne pôsobenie tepla v rozmedzí 42–45 °C neslúži na okamžitú deštrukciu, ale na „uvoľnenie“ štruktúry zhlukov. Tým sa stávajú prístupnými pre prirodzený glymfatický systém mozgu – akési „nočné upratovanie“, počas ktorého nervová sústava počas spánku vyplavuje metabolický odpad. Vďaka tomuto procesu sa kedysi odolné patologické agregáty stávajú pre organizmus spracovateľnými a môžu byť bezpečne eliminované. V laboratórnych podmienkach in vitro toto teplo narúša jemné vodíkové väzby v β-skladaných štruktúrach. Ide o experimentálny prístup zatiaľ overovaný na bunkových kultúrach a zvieracích modeloch — klinické aplikácie u ľudí sú vo veľmi ranom štádiu výskumu.
- Dezintegrácia a čistenie: Po narušení stability sa priónový agregát mechanicky rozpadne na menšie, rozpustnejšie časti. Tie sa stávajú pre prirodzené čistiace mechanizmy mozgu (mikroglie) ľahko rozpoznateľným odpadom, ktorý dokáže organizmus konečne odbúrať.
Hoci ide o experimentálne metódy, ktoré sú zatiaľ vo fáze výskumu in vitro a testov na zvieracích modeloch, predstavujú zásadný posun. Namiesto toxických chemických zásahov sa tak otvára cesta k „molekulárnej chirurgii“ bez použitia skalpela.
Je dôležité rozlišovať medzi patologickým zhlukom a následným poškodením tkaniva. Zatiaľ čo zhluky priónov sú aktívnym patogénom, vakuoly (diery v tkanive) sú už len smutným jazvami po mŕtvych neurónoch. Cieľom fyzikálnej dezintegrácie pomocou nanočastíc je zasiahnuť patogén skôr, ako stihne neurón zničiť. Úspešná liečba preto závisí od včasnej diagnostiky, ktorá nám umožní rozbiť agregáty v čase, keď je nervová bunka ešte schopná regenerácie a následného očistného procesu.
Limity súčasných prístupov: Napriek sľubným výsledkom laboratórnych experimentov zostáva využitie nanotechnológií pri liečbe priónových ochorení zatiaľ vzdialené klinickej praxi. Najväčšou výzvou je bezpečné doručenie nanočastíc cez hematoencefalickú bariéru do presne definovaných oblastí mozgu a zároveň zabezpečenie, aby sa zahrievali iba patologické agregáty bez poškodenia okolitého nervového tkaniva. Ďalším problémom je veľmi neskorá diagnostika priónových ochorení – v čase objavenia prvých príznakov býva časť neurónov už nenávratne poškodená. Súčasný výskum preto smeruje k vývoju novej generácie inteligentných nanočastíc, citlivejších diagnostických metód a kombinovaných terapií, ktoré by dokázali patologické proteíny odhaliť a odstrániť ešte pred vznikom rozsiahleho poškodenia mozgu.
Záver
Objav priónov patril k najväčším prekvapeniam modernej biológie. Ukázal, že biologická informácia nemusí byť uložená iba v génoch. Niekedy stačí samotný tvar molekuly.
To, čo sa kedysi zdalo nemožné, dnes predstavuje jeden z najdôležitejších kľúčov k pochopeniu neurodegeneratívnych ochorení. Prióny nám pripomínajú, že aj po desaťročiach intenzívneho výskumu dokáže príroda stále prekvapiť – a že hranica medzi zdravím a chorobou môže byť ukrytá v jedinom nesprávnom záhybe proteínu.
Dnes vieme, že podobné mechanizmy nesprávneho skladania proteínov zohrávajú úlohu aj pri bežných ochoreniach, ako je Alzheimerova a Parkinsonova choroba. Prióny tak prestali byť len medicínskou kuriozitou a stali sa kľúčom k porozumeniu širších neurodegeneratívnych procesov.
Zoznam použitej a odporúčanej literatúry
- Prusiner, S. B. (1998). Prions. Proceedings of the National Academy of Sciences of the United States of America, 95(23), 13363–13383. (Základný prehľadový článok od nositeľa Nobelovej ceny)
- Prusiner, S. B. (2013). Prion Biology and Diseases. Cold Spring Harbor Laboratory Press. (Komplexná monografia, jedna z najautoritatívnejších)
- Collinge, J. (2001). Prion diseases of humans and animals: Their causes and molecular basis. Annual Review of Neuroscience, 24, 519–550.
- Aguzzi, A., & Polymenidou, M. (2004). Mammalian prion biology: One century of evolving concepts. Cell, 116(2), 313–327.
- Weissmann, C. (2004). The state of the prion. Nature Reviews Microbiology, 2(11), 861–871.
- Sim, V. L., & Caughey, B. (2009). Recent advances in prion biology. Current Opinion in Structural Biology, 19(1), 1–7.
- Wadsworth, J. D. F., & Collinge, J. (2011). Molecular basis of human prion disease. Acta Neuropathologica, 121(2), 171–185.
- Head, M. W., & Ironside, J. W. (2012). Review: Creutzfeldt-Jakob disease and related human transmissible spongiform encephalopathies. Neuropathology and Applied Neurobiology, 38(1), 1–15.
- Mallucci, G., & Collinge, J. (2005). Rational targeting for prion therapeutics. Nature Reviews Neuroscience, 6(1), 23–34.
Ďalšie odporúčané zdroje pre hlbšie pochopenie:
- Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science, 216(4542), 136–144. (Historicky dôležitý pôvodný článok)
- Dobson, C. M. (2003). Protein folding and misfolding. Nature, 426(6968), 884–890. (Kontext nesprávneho skladania proteínov)
- Soto, C. (2012). Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience, 13(1), 49–60. (Prepojenie na Alzheimerovu a Parkinsonovu chorobu)





